ABSTRACT
Introduction: We have previously demonstrated that the gut microbiome metabolite trimethylamine (TMA) exerts significant hemodynamic effects, notably a pronounced hypertensive response [1]. While the Trace Amine-Associated Receptor 5 (TAAR5) is a known molecular target for TMA, its physiological role has been studied almost exclusively within the central nervous system [2], with its peripheral cardiovascular functions remaining unexplored. Furthermore, it remains unknown whether TMA-induced cardiovascular responses differ in the setting of established hypertension.
Aims: To investigate the dose- and strain-dependent hemodynamic responses to intravenous TMA in normotensive Wistar-Kyoto (WKY) and Spontaneously Hypertensive Rats (SHR), and to determine if these cardiovascular effects are mediated by the TAAR5 receptor.
Methods: WKY and SHR rats (n=5-6 per strain and dose) received intravenous TMA at doses of 45, 135, and 405 µmol/kg. Blood pressure (BP) was continuously monitored for 20 minutes under urethane 1.5 g/kg i.p. anaesthesia. To assess the mechanistic role of TAAR5, a subset of WKY rats was pretreated with a TAAR5 inhibitor (Z1230215228, Enamine, equimolar dose, i.v.) prior to the 135 µmol/kg TMA challenge. Baseline TAAR5 mRNA expression was quantified via RT-qPCR in the heart (right and left ventricle), kidney (medulla and cortex), jejunum, colon, and liver of both strains. All procedures accorded with Directive 2010/63/EU and were approved by Local Bioethical Committee.
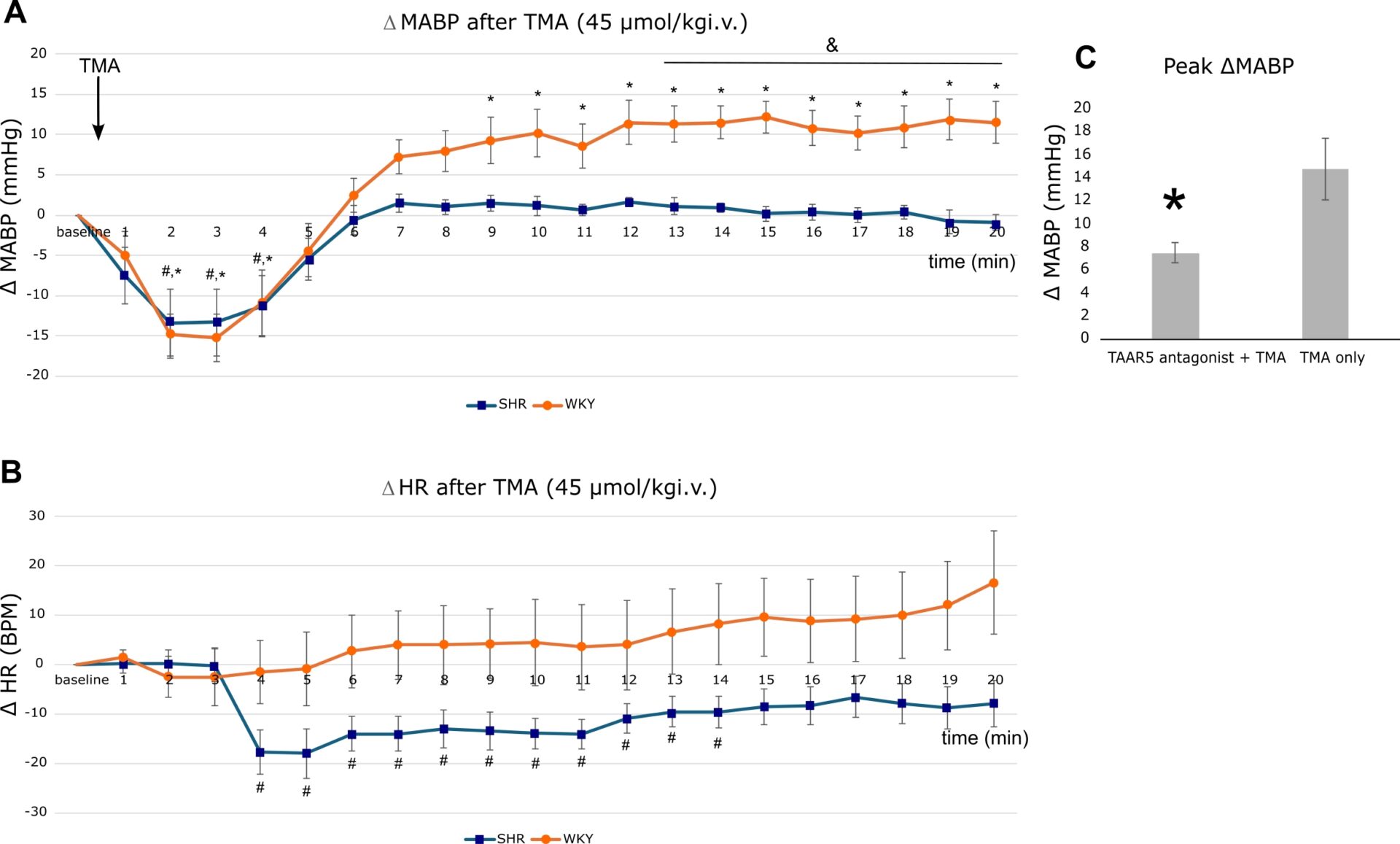
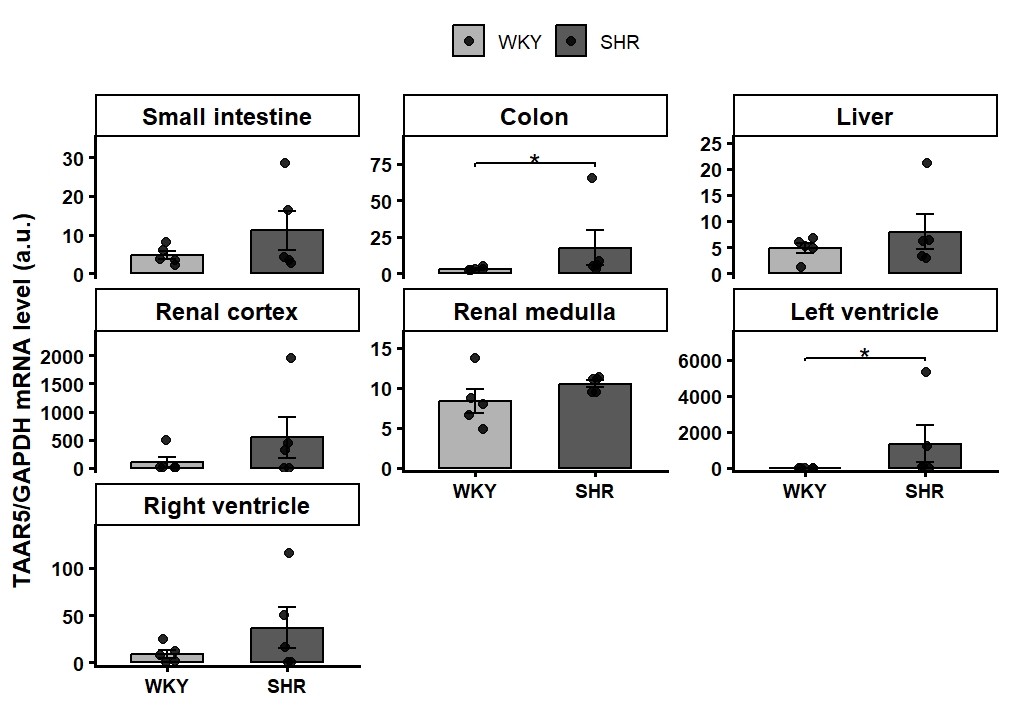
Results: At the lowest TMA dose (45 µmol/kg), rats exhibited a distinctly strain-dependent hemodynamic response (Two-way mixed ANOVA, Time × Strain interaction, p<0.001, Figure 1A,B). While both strains displayed a biphasic BP reaction, the subsequent hypertensive rise was significantly less pronounced in SHR compared to WKY (BP WKY vs SHR p<0.05 from minute 13 through minute 20 post-injection, independent t-tests, Holm-Bonferroni corrected). Concurrently, TMA induced a significant drop in HR in SHR at both 45 and 405 µmol/kg doses, whereas HR in WKY rats remained unchanged (Two-way mixed ANOVA, Time × Strain interaction, p<0.001). At higher doses (135 and 405 µmol/kg), the temporal BP trajectories did not significantly differ between strains. Mechanistically, pretreatment with the TAAR5 inhibitor in WKY rats significantly attenuated the TMA-induced hypertensive response (Peak MABP within the first 10 min: 7.50±0.88 vs. 14.78±2.64 mmHg, p=0.03, Welch’s t-test, Figure 1C). Tissue profiling revealed ubiquitous TAAR5 expression across all studied organs; notably, baseline TAAR5 expression was significantly upregulated in the left ventricle and colon of SHR compared to WKY rats. (Figure 2).
Conclusion: TMA elicits a dose-dependent, biphasic hemodynamic response, with the pressor phase being significantly blunted in hypertensive subjects, likely driven by a strain-specific bradycardic reflex. In normotensive rats, this TMA-induced pressor effect is mediated by the TAAR5 receptor, providing the first direct evidence of its peripheral cardiovascular function. Furthermore, the targeted upregulation of TAAR5 in the left ventricle and colon of SHR offers a plausible molecular basis for their altered reflex, pointing toward a disease-specific remodeling of the microbiome-cardiovascular axis in hypertension. However, further functional studies in hypertensive models are warranted to confirm this direct mechanistic link.