Introduction
TDP-43 proteinopathies, a group of neurodegenerative diseases encompassing the amyotrophic lateral sclerosis/frontotemporal dementia (ALS/FTD) spectrum, are characterised by marked oligodendrocyte dysfunction; however, the mechanistic contribution of these cells to neurodegeneration remains unclear. Under physiological conditions, TDP-43 is predominantly nuclear and regulates RNA processing, transport, and stability. In the majority of ALS/FTD cases, TDP-43 mislocalises to the cytoplasm, resulting in both loss of nuclear function and toxic gain of function. Accumulating evidence implicates oligodendrocyte pathology in ALS/FTD. Highly myelinated axons are particularly vulnerable to degeneration, and cytoplasmic TDP-43 aggregation is observed in both neurons and oligodendrocytes, with a substantial subset of patients exhibiting predominant oligodendroglial pathology. Moreover, patient-derived oligodendrocytes are neurotoxic in vitro, and oligodendrocyte-specific disruption of ALS-associated genes induces axonal damage in vivo. Despite this evidence, most studies have focused on neuronal cytoplasmic TDP-43 expression, leaving the contribution of oligodendrocytes relatively underexplored.
Aims/Methods
To investigate the impact of cytoplasmic TDP-43 expression on the CNS from neurons and oligodendrocytes, we generated a series of novel humanised zebrafish models for in vivo analysis. In these models, the endogenous zebrafish TDP-43 gene is replaced with human TDP-43 carrying the Δnls mutation, which drives cytoplasmic mislocalisation. Humanised lines express cytoplasmic TDP-43 at physiologically relevant levels either ubiquitously (∆nlsubi), neuron-restricted (∆nls neuro), or restricted to the oligodendrocyte lineage (∆nlsoligo). All regulated zebrafish procedures were sub threshold severity and in accordance with the project licence protocols. 2- anova was conducted for each experiment for significance with at least 5 or more individuals pooled from 3 independent clutches.
Results
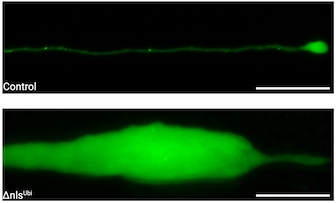
Ubiquitous expression of cytoplasmic TDP-43 resulted in early dysfunction of both neurons and oligodendrocytes, followed by lethality at approximately 10 days post-fertilisation (dpf). Analysis of myelinated upper motor neurons revealed pronounced distal swellings at 5dpf, which preceded motor neuron degeneration several days later. At 5dpf, oligodendrocyte numbers were reduced by ~30%, while myelin levels were decreased by ~60%.
Restricting cytoplasmic TDP-43 expression to neurons similarly caused severe distal axonal swelling and degeneration of myelinated upper motor neurons, although onset was delayed by ~2 days. Notably, oligodendrocyte numbers remained unchanged in the ∆nlsneuro; however, global myelin levels were reduced by ~35%, indicating impaired oligodendrocyte function. Preventing myelination via Olig2 knockdown directly in the ∆nlsneuro line exacerbated axonal swellings, suggesting that residual myelin is neuroprotective rather than neurotoxic.
In a similar fashion, oligodendrocyte-restricted cytoplasmic TDP-43 expression did not alter oligodendrocyte numbers but significantly reduced myelin levels by 5dpf. Of note, analysis of highly myelinated upper motor neurons revealed distal axonal structural abnormalities, indicating that cytoplasmic TDP-43 expression exclusively in oligodendrocytes disrupts axonal integrity in a non-cell-autonomous manner.
Conclusions
These findings demonstrate that cytoplasmic TDP-43 expression in either neurons or oligodendrocytes is sufficient to induce axonal dysfunction and reduce myelin levels. In ∆nlsneuro, myelin reduction precedes axonal degeneration, and inhibition of myelination exacerbates axonal pathology. Furthermore restricting cytoplasmic TDP-43 to the oligodendrocyte lineage also causes a reduction in myelin levels and a disruption to axonal integrity. Taken together, this suggests a pathological feedback loop between neurons and oligodendrocytes, underscoring the reciprocal contributions of neurons and oligodendrocytes to ALS/FTD pathogenesis.